科技日报记者 张梦然
英国“深度思维”(DeepMind)公司日前公布了新一代“阿尔法折叠”(AlphaFold-latest),不仅准确性显著提高,预测范围还从蛋白质扩展到其他生物分子,包括配体。该模型已可以预测蛋白质数据库(PDB)中的几乎所有分子,预测精度可以达到原子级。
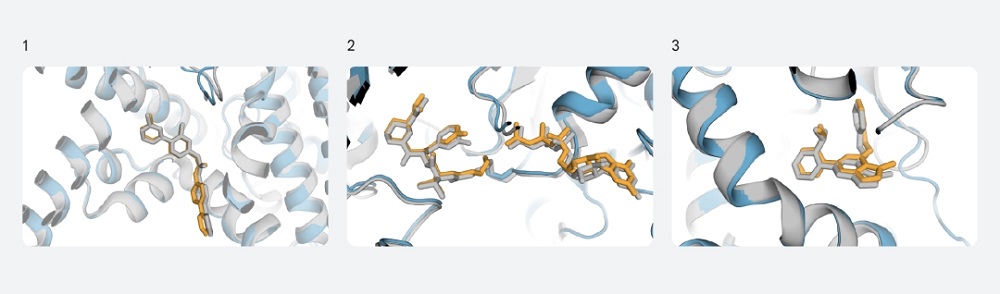
对抗癌分子的结合(PORCN)、关键癌症靶标的共价配体结合(KRAS)以及脂质激酶变构抑制剂的结构预测,新模型预测部分为彩色,案例实验预测部分为灰色。图片来源:“深度思维”官网
“深度思维”公司表示,这一模型的扩展功能和性能将加速生物医学领域的突破,推动人类迈向下一个“数字生物学”时代,为疾病通路的功能研究、基因组学、生物可再生材料、植物免疫、潜在治疗靶点、药物设计机制、蛋白质工程和合成生物学等领域提供了前所未有的见解,并打造了一种全新的平台。
此次,研究人员展示了新一代模型在预测蛋白质折叠之外的精确结构方面的突出能力,它可以对配体、蛋白质、核酸和翻译后修饰进行高度精确的结构预测。这意味着它在抗体结合预测领域会表现突出;在与药物发现相关的蛋白质结构问题上,其明显超越“前辈”和行业标准;该模型还具备联合建模所有原子位置的能力,能够更全面地揭示蛋白质和核酸与其他分子相互作用时的灵活性。
传统方法需要使用刚性蛋白质结构和对接方法来预测蛋白质—配体结构,而新一代模型无需这些先验信息,反而表现出更高准确性。可以说,其重新定义了预测蛋白质—配体结构的标准,使得以前未知结构的蛋白质也可以被预测。
新一代模型将应用于治疗药物设计,帮助快速、准确地描述对治疗疾病非常重要的多种类型的大分子结构。此外,经过蛋白质、配体、核酸以及翻译后修饰结构的模拟解锁,该模型可以为基础生物学研究提供更迅速且准确的工具。
总编辑圈点:
人体每一种蛋白质都包含几十到几百种氨基酸,氨基酸的顺序决定了它们之间的作用,赋予蛋白质复杂的三维形状,进而决定了蛋白质的功能。几十年来,科学家利用X射线晶体学或低温电子显微镜等实验技术来破译蛋白质的三维结构,但这种方法可能需要数月甚至数年,且未必见效。“阿尔法折叠”的出现被认为改变了游戏规则,取得了根本性突破,同时,我们欣喜地看到这一AI还在不断升级、扩展,以更高的准确率覆盖到了更多的复合物。





